


Figure 1. Process Flow of Biopharmaceutical Manufacturing
Continuing from our last post, ‘Biopharmaceutical Manufacturing 1‘, this current post, ‘Biopharmaceutical Manufacturing 2‘, elaborates upon the steps in biopharmaceutical manufacture after insertion of the ‘gene of interest‘ into the ‘Expression Vector‘. We therefore begin this post at Step 6 of our Manufacturing Process Flow in which the ‘Vector‘ transports our gene of interest into the host cell. This is presented in even greater detail below in Figure 2.
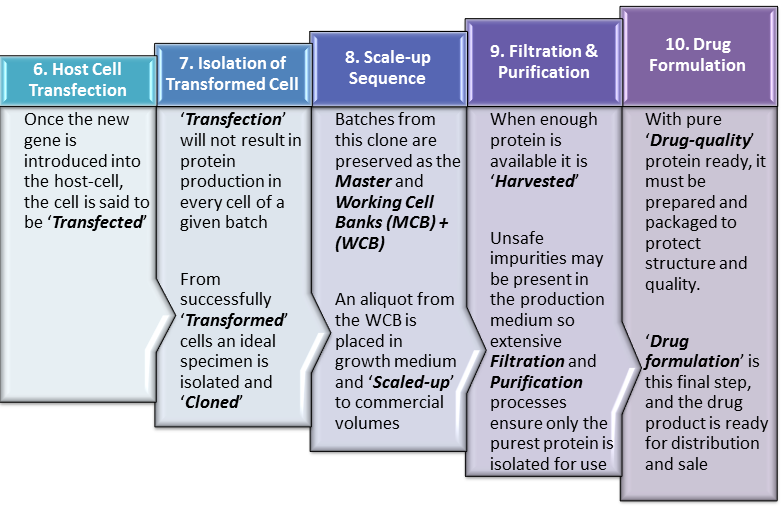
Figure 2. Final Steps in Biopharmaceutical Manufacture
Click to review Process Flow
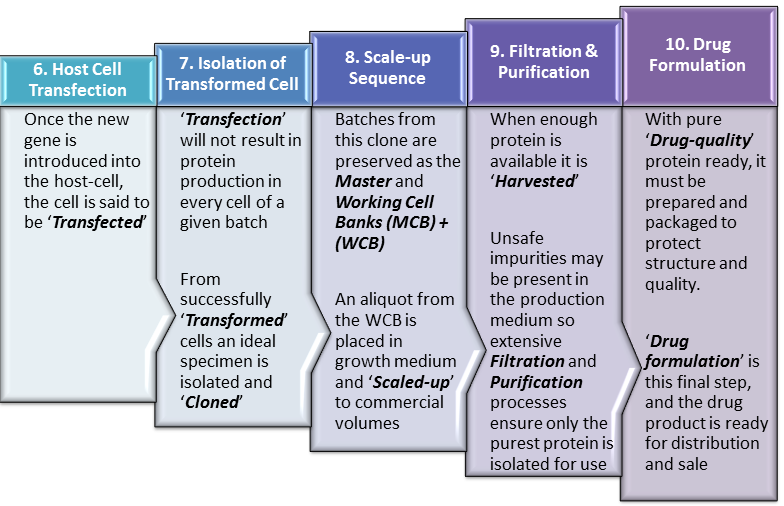
Step 6. Host Cell Transfection
Having successfully integrated the ‘gene-of-interest‘ into our vehicle (Vector), methods are further employed to get the vector into the host-cell, effectively delivering the gene. Plasmids and viruses can achieve cell entry quite readily but even more involved methods can also be employed such as direct injection of DNA into the cell.
At this point, the specific DNA sequence (gene) for our protein has gained entry to the host cell and ideally becomes a part of the host-cell’s own complement of genetic material, otherwise known as its ‘Genome‘. At this point the host cell should be replicating the human gene as it undergoes the normal process of cell division. More importantly for the manufacturer’s purposes, the host-cell should also start producing copies of the the human protein along with the usual host-cell proteins it normally produces.
Click to review Process Flow Click to review Initial Steps
Step 7. Isolation of Transformed Cells
While the ideal is for every cell into which a vector enters, to be ‘Transformed‘ producing human protein without exception, this is not the case in reality. What actually obtains is that many of the cells into which the vector enters, for various reasons do not in fact become active or efficient producers of the human protein. Some will transform but produce relatively small amounts of the human protein while others may produce defective or low quality human protein. These non-productive or low-productivity cells are not useful to the manufacturer and must therefore be identified and separated from useful cells which produce large amounts of high-quality protein.
Share this Post
Differentiation between cells producing and not producing the desired protein can be achieved using labelled-antibodies specific to the protein of interest. These labels can include fluorophores, chromophores or radio isotopes among others.
Once the highest performing cells are identified, bench-marking assays may be conducted to see which cells perform best (e.g. produce protein at the highest rate). After this, a single high-performing cell is usually selected and placed in growth medium that promotes replication of the cell in large numbers. Because this batch would have been inoculated from a single cell (or colony) all the new cells created will be identical (clones) possessing the same ideal characteristics.
Samples of this inoculum are then partitioned, frozen in liquid nitrogen and stored as the ‘Master Cell Bank‘ (MCB). If problems ever arise with day-to-day production through contamination of the production mixture as one example, aliquots from the MCB can be taken to restart production with the purest of cells ensuring continuity of process.
Typically only a relatively small amount of the inoculated growth mixture goes into deep frozen storage as the MCB. The rest is stored for daily usage in production runs. This portion used for daily production is called the ‘Working Cell Bank‘ (WCB).
Click to review Process Flow Click to review Initial Steps
Step 8. Scale-up Sequence
 The size of aliquots required from the WCB to start a new production run is typically quiet small. These aliquots are used to inoculate a very small amount of growth medium in a laboratory setting. These batches of growing cells must gradually be transferred to larger and larger vessels of growth medium to progressively scale the production process up to a commercial level. As such, the process may start with only a few liters of growth medium but end in bioreactors of up to 1000 liters.
The size of aliquots required from the WCB to start a new production run is typically quiet small. These aliquots are used to inoculate a very small amount of growth medium in a laboratory setting. These batches of growing cells must gradually be transferred to larger and larger vessels of growth medium to progressively scale the production process up to a commercial level. As such, the process may start with only a few liters of growth medium but end in bioreactors of up to 1000 liters.
Click to review Process Flow Click to review Initial Steps
Step 9. Filtration & Purification
 Once a predefined biomass volume has been reached or enough human protein has been produced in the bioreactors, it must be harvested. To achieve this it must be separated from all other proteins and impurities found in the growth mixture. These impurities and contaminants can include proteins, waste-products from the growth process, cellular, bacterial or even viral fragments among others.
Once a predefined biomass volume has been reached or enough human protein has been produced in the bioreactors, it must be harvested. To achieve this it must be separated from all other proteins and impurities found in the growth mixture. These impurities and contaminants can include proteins, waste-products from the growth process, cellular, bacterial or even viral fragments among others.
Initially, courser filtration methods remove large particles and fragments but as the process progresses the purification mechanisms become more sophisticated ensuring removal of ever smaller contaminants. Multiple purification runs using varied techniques such as ultrafiltration, numerous forms of chromatography such as gel filtration chromatography and ion exchange chromatography may be employed. By the end of this process even microscopic impurities are reduced to a minimum leaving behind only the purest human protein, now called the Active Pharmaceutical Ingredient (API).
Click to review Process Flow Click to review Initial Steps
Step 10. Drug Formulation
Once the pure API has been isolated, it must be appropriately stabilized and packaged to prevent breakdown and molecular damage on its way to the final user. This is achieved through the addition of excipients and different forms of physical processing such as freeze-drying all of which will enhance molecular stability. The product is then packaged in a syringe, vial or ampoule as may be appropriate and is ready for distribution to the final customer usually under refrigerated or other highly-controlled conditions.
Conclusion
Biopharmaceutical Manufacturing Process Flow
As noted in the opening lines of ‘Biopharmaceutical Manufacturing 1‘, the process of biopharmaceutical manufacturing is a highly complex, multistage process highly prone to variability because it is based in living cells. This means that differences in factors such as cell number (or concentration), oxygen levels, carbon dioxide levels, temperature, pH, nutrient concentrations and availability, pressure, flux rate, waste substances among many others can all result in changes to the final product. It is therefore a significant challenge to maintain full control over the production process of a given biopharmaceutical and even more so to attempt to replicate it. Any attempt to achieve full control over the process requires meticulous documentation and record-keeping to identify and manage the potential sources of variability.
The complicated mechanisms of action of biopharmaceuticals make predicting the full spectrum of their clinical effects unlikely. This is further complicated by their large size and complex structure. This complexity further translates to the production process as well. In fact a defining characteristic of biopharmaceuticals, different from conventional small-molecule drugs is that the clinical effect of production process changes cannot be readily predicted.
In practical terms this means changes anywhere in the process, planned or otherwise, can result in alterations to the final product that may have clinical significance. This possibility dictates that where attempts are made to produce copies of biopharmaceuticals and even when modifying the production process of an existing product, detailed assessments must be made throughout.
In the specific instance where a second manufacturer decides to make a copy of an existing biopharmaceutical product, detailed information on the existing manufacturing process is likely to be unavailable. In such cases differences between the two products will exist and consistent with both WHO and EMA recommendations, head-to-head clinical trials will be needed to specifically to assess the clinical impact of those differences. This requirement when making copies of biological products, for comparative clinical trials outside the scope of bioequivalence studies is a defining difference between them and small molecule drugs.
CLICK HERE to view Downloads on Pharmaceutical Manufacturing from CaribbeanBiopharma