

The Critical Importance of the Master Cell Bank


Figure 1. Cell Banks are stored stably in liquid nitrogen tanks at -150 degrees Celsius
Our last post, ‘The Product is the Process – The Master Cell Bank 1‘, unequivocally presented the genetically-engineered cells that produce ‘protein-based drugs’, as the foundation of biopharmaceutical production. So fundamental are these cell lines in fact, that if a manufacturer were to lose them to contamination for example, further production would be impossible. Additionally, restoration of supply would entail the daunting task of recreating the Master Cell Bank or MCB, a feat which is not likely to be achieved irrespective of how well documented the previous process was. (See Part 1 for a detailed explanation of the MCB). This is because, so many variables are involved, that repeating the process with such precision as to yield an identical MCB to the one lost, is considered essentially unattainable.
As such, even after creating version 2 of the MCB, the manufacturer would be required to undertake a detailed comparability exercise to prove that the product generated by the new cell line, is sufficiently similar to that produced by the old so as to be treated equally. Only after demonstrating the required level of similarity could the existing marketing authorization / license be applied to the new product. The comparability exercise would undoubtedly include completing new clinical trials as well, because the structural complexity of biopharmaceuticals is typically not amenable to comparison by physico-chemical means only. As such, demonstrating an ‘adequate’ level of similarity where it matters most, at the clinical level, can be definitively proven for biopharmaceuticals, only through clinical trials designed to test safety, efficacy and immunogenicity which is the ability to elicit responses, often negative ones, from a patient’s immune system.
This recognition therefore underpins the principle that, ‘If the cell line is not identical, then the product can’t be either!’
Furthermore, the point of the exercise would not be proving ‘standalone efficacy or safety of the new product‘, but rather, proving that ‘the data supporting the existing license (of the old product), also applies to the new‘. The goal of the exercise would therefore be comparative safety and efficacy, comparing new to old. Accordingly, the various tests and studies required should be comparative and head-to-head in nature. In other words, each study should consist of parallel arms assessing each parameter of interest one with the new drug, the other with the old drug simultaneously.
The Unsuccessful Comparability Exercise
In the doubly unfortunate scenario where our manufacturer having lost its cell line, is also unable to prove the requisite level of similarity between old and new versions of its product, the organization may find itself facing the nightmare scenario of developing the same drug all over again ‘from scratch’. This means assuming the cost and complexity of both a full pre-clinical and clinical trial programme, as opposed to what should have been the detailed, yet abbreviated requirements of the simpler and less costly comparability exercise.
That being the case, only after completion of what would be its second, ‘full’ development programme, could the manufacturer submit the accumulated data from these new studies and trials as part of its application for marketing authorization. Given the circumstances however, the manufacturer will be submitting the ‘Version 2‘ molecule as a New Molecular Entity (NME) on its own merit, detached from the previous marketing authorization and the data supporting it.
The need for such robust assessment of similarity, attests to just how unlikely the authorities themselves consider the possibility of developing a cell line that is identical to any other and by extension, replicating the process that yields a biopharmaceutical that is identical to any other. It is worthwhile to emphasize that this requirement applies even to attempts by the same manufacturer to develop a cell line identical to previous ones of its own design. This is quite remarkable given that the manufacturer has full access to documentation and details of the development process of the original cell line. As a result, it might otherwise have been assumed that with such records the cell line could easily be replicated, but as we saw above, not so!
This recognition therefore underpins the principle that, ‘If the cell line is not identical, then the product can’t be either!‘ Producing multiple batches of near-identical product, therefore starts with using cells from the same ‘one-of-a-kind’ cell line (MCB) and then further ensuring that the conditions under which these cells grow and produce the protein of interest, are as near-identical as possible from one batch to another.
The Myozyme / Lumizyme Case
We have a number of times, stated that biopharmaceutical production is both very sensitive as well as highly prone to variation, but nothing makes the case quite as convincingly as an example. So to highlight just how sensitive the process can be and the impact of product changes on marketing authorization, we now explore a case from the Boston-based biotech firm, Genzyme Corporation. Genzyme’s product Myozyme® is a protein-based, enzyme-replacement medicine for treatment of the rare genetic disorder, Pompe Disease. The disorder results from a genetically-derived deficiency in the enzyme, ‘acid alpha-glucosidase‘ which is necessary for breakdown of glycogen to glucose before it can be used for energy in the body. As would be expected, this affects the body’s management of its energy reserves and has a particularly detrimental effect on skeletal and cardiac muscle.
Share this Post
Genzyme received initial approval from the US Food and Drug Administration (FDA) for use of Myozyme® to treat Pompe Disease in April 2006 based upon production in 160-liter bioreactors at its original facility. Increasing demand for the treatment of this often fatal disorder, prompted Genzyme to increase production. This lead to the commissioning of a new 2000-liter production facility and this is where things got interesting; confirming just how complex and unpredictable biopharmaceutical production can be.
Despite all other variables remaining the same, the only obvious difference being the size of the bioreactors, it turns out that the profile of carbohydrate side-chains found on the protein from the new facility, was different from that of the protein produced in the original 160-liter facility. Sufficiently different in fact, that the FDA denied extending the existing Biologics License Application (BLA) of Myozyme® to product from the new facility. As such, Genzyme was forced to apply for a separate BLA for the product from the new facility, effectively licensing what should have been Myozyme®, as a new drug. Under the new BLA, the product from the 2000-liter facility had to carry a distinct brand name from the old despite both drugs containing the protein alglucosidase alfa. Hence, Lumizyme® was born, supported by additional clinical trials to substantiate its safety and efficacy. In 2010, Lumizyme® received limited approval for treatment of (non-infantile) Late Onset Pompe Disease (LOPD) in patients who are 8 years of age and older, pending further data from Genzyme. Having received the requested data, the FDA later expanded Lumizyme’s approved indications to match those of Myozyme® in 2014, covering both treatment of patients with LOPD and infantile-onset disease in patients younger than 8 years of age . While both products have the same active pharmaceutical ingredient (API), alglucosidase alfa, the same indications and are derived from the same cell line, they continue to hold separate BLAs and are marketed under separate brand names today.
It is noteworthy, that Genzyme founded in 1981, was at the time of this incident, a well established pioneer in pharmaceutical biotechnology and therefore an experienced operator in biopharmaceutical production. Despite Genzyme’s wealth of organizational experience and expertise they did not anticipate the hurdles they would be forced to overcome. This case in particular, underlines just how subtle changes to the biopharmaceutical production process can be and yet lead to changes profound enough to obstruct marketing authorization.
The Cost of Losing the Master Cell Bank
Our Myozyme® / Lumizyme® example highlighted how burdensome changes to the biopharmaceutical production process can be, but importantly the consequences of these changes more often than not, are also accompanied by economic implications.
As far back as 2005, Tonkens estimated that drug development costs were in the range of $1.8-1.9 billion per pharmaceutical product. DiMasi, Hansen and Grabowski (2003) earlier estimated that pre-clinical costs accounted for only about 30% of the total, suggesting that the majority of development expenditures are associated with the human phase of testing (i.e. Clinical Trials). As noted in the article by Booth (2014), ‘A Billion Here, A Billion There: The Cost Of Making A Drug Revisited‘, more recent estimates have ‘upped’ the total development figure to $2.5 billion, with the proportion of pre-clinical to clinical costs remaining largely the same.
If these estimates are correct, the clinical development programme alone, for the average drug, could cost in the region of $1.7 billion. The unfortunate manufacturer that loses its cell stock is therefore likely to be faced with a shock similar, if not worse than Genzyme’s earlier predicament. This could mean reinvesting, hundreds of millions of dollars just to prove that the second version of their cell stock, provides a comparable product to the first.
Protecting the Master Cell Bank
In light of such risk, ensuring the integrity of their cell lines once developed, is of paramount importance for any manufacturer. For this reason, biopharmaceutical companies typically use a ‘Two-tiered Cell Banking System‘ as illustrated in Figure 2 below, to insulate the all-important Master Cell Bank from damage and depletion. By using this system, production capacity can be maintained almost indefinitely without ever significantly depleting or placing at risk, the stock of original cells created for the purpose.
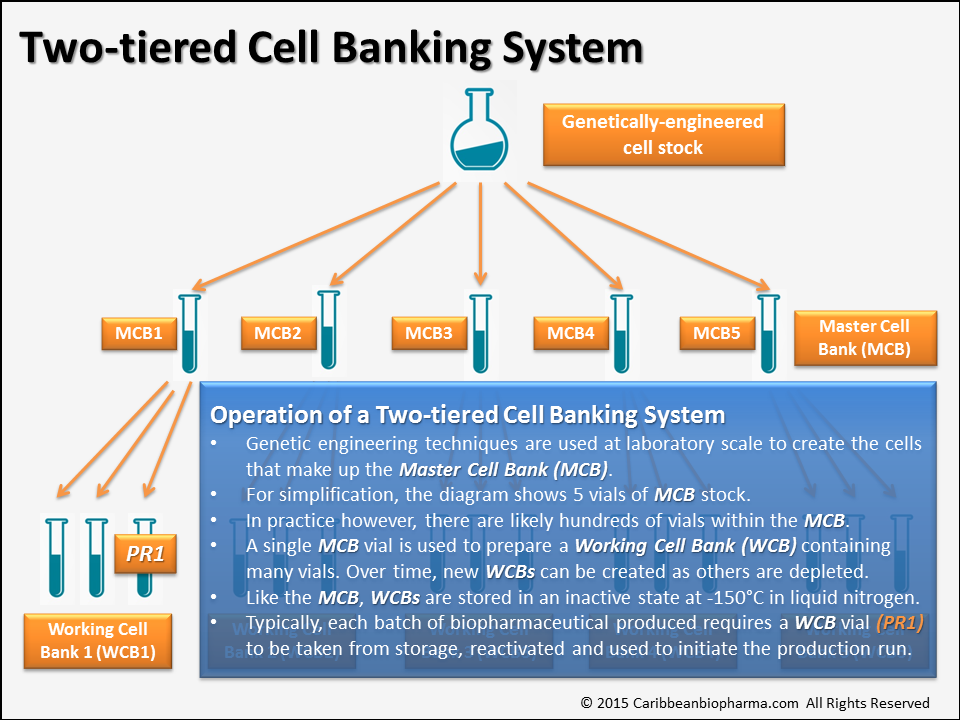
Figure 2. Diagram illustrating the operation of a two-tiered Cell Banking System
As detailed above, the pivotal role of the Master Cell Bank means that daily interaction with it, would pose an unacceptable risk for any manufacturer. As such, portions of the MCB are taken-off and stored separately for ‘day-to-day‘ use as the feed stock for each production run, soon after creation. These separated ‘day-to-day‘ working batches of cells, unsurprisingly are called Working Cell Banks or WCBs and while portions of the WCBs may be accessed every day, the remaining stock of both the MCB and WCB are very securely stored away in separate storage tanks. While the WCBs are accessed on a regular basis, maybe daily, the MCB is essentially left untouched as a reserve in the event the WCBs are depleted or compromised. Both the MCB and WCBs are stored stably in liquid nitrogen at -150 degrees Celsius protecting them over the drug’s entire life-cycle.
Conclusion
In this final part of our two-piece article on how a company’s cell stock and Master Cell Bank (MCB) specifically, can profoundly impact biopharmaceutical production, we looked at not only the practical importance, but also the economic importance of protecting the MCB and how to do so. The Myozyme® / Lumizyme® example illustrated just how subtle the differences that impact production are and equally, how profound the impact on the final product can be.
It is important to note that despite all the effort placed on highlighting how easily changes to the product can occur, that we also recognize that not all changes to the product will be clinically significant. In fact, the majority of changes may well be of no importance at the clinical level. Despite this, the critical point is that ‘Safety should not be assumed‘. In the context of very serious diseases, which by the way is the setting in which biopharmaceuticals are most often used, ‘Patient safety should always be of supreme importance‘. This should prompt manufacturers to do their absolute best in identifying not similarities, but differences between product batches and product versions where applicable.
Additionally, in the context of biopharmaceuticals whose complete structures cannot be fully characterized through physico-chemical means only, clinical trials and most often, head-to-head comparative clinical trials are the best bet for determining what effect product changes will have on patients.
In the final analysis, both manufacturers and regulators must keep in very sharp focus, that the highest regard needs to be given to the manufacturing process because ultimately as described before, ‘The Product is the Process‘
References
1. Booth, B. (2014) A Billion Here, A Billion There: The Cost Of Making A Drug Revisited [Online] LifeSciVC.com. Available from: http://lifescivc.com/2014/11/a-billion-here-a-billion-there-the-cost-of-making-a-drug-revisited/ (Accessed: 24 May 2015)
2. DiMasi, Hansen, Grabowski (2003), ‘The price of innovation: New estimates of drug development costs’, J. Health Economics 2003, pp. 151-185
3. FDA (2006) FDA News Release: FDA Approves First Treatment for Pompe Disease [Online]. (Accessed: 3 Jun 2015)
4. FDA (2010) FDA News Release: FDA Approves New Treatment for Late-Onset Pompe Disease [Online]. (Accessed: 3 Jun 2015)
5. FDA (2014) FDA Expands approval of drug to treat Pompe Disease to patients of all ages; removes risk mitigation strategy requirements [Online]. (Accessed: 3 Jun 2015)
6. Taylor, N. (2009) Myozyme becomes Lumizyme after biologics scale-up [Online] Montpelier, France: In-Pharma Technologist.com. Available from: http://www.in-pharmatechnologist.com/Ingredients/Myozyme-becomes-Lumizyme-after-biologics-scale-up (Accessed: 3 Jun 2015)
7. Tonkens, R. (2005), ‘An overview of the drug development process’, Physician Executive, 31 (3), pp. 8
One Comment on “The Product is the Process – The Master Cell Bank 2”
This is a great article , very informative. Thanks for your research and insight.